





Abstract
Glutathione transferase zeta (GSTZ1-1) catalyzes the isomerization of maleylacetoacetate (MAA) to fumarylacetoacetate, the penultimate step in the tyrosine degradation pathway. GSTZ1-1 is inactivated by dichloroacetic acid (DCA), which is used for the clinical management of congenital lactic acidosis and is a drinking-water contaminant. Metabolic changes associated with chemically induced GSTZ1-1 deficiency are poorly understood. The objective of this study was to investigate the biochemical and toxicological effects of giving 0.3–1.2 mmol DCA/kg/day for 5 days on MAA-metabolism in male Fischer rats. Urine from DCA-treated rats inhibited δ-aminolevulinic acid dehydratase (δ-ALAD) activity, which is used for the diagnosis of hereditary tyrosinemia type I. Mass spectrometric analyses of urine from rats given DCA demonstrated elevated excretion of MAA and its decarboxylation product, maleylacetone (MA); succinylacetone (SA), the reduced analogue of MA, was not detected. DCA-induced changes in MA excretion were dose-dependent and were significantly elevated after day 2 of treatment. MA excretion was reversible after discontinuation of DCA treatment and was enhanced 10-fold by the coadministration of homogentisic acid (HGA). MA was cytotoxic to hepatocytes in vitro (EC50 ~ 350 μM) but morphological changes were not observed in liver, kidney, and brain of rats given both DCA and HGA. These data indicate that DCA-induced inactivation of GSTZ1-1 leads to formation of an MAA-derived intermediate, MA, that may be a mediator and biomarker for DCA-associated toxicities.
Tyrosine is a nonessential amino acid that is derived from the diet or from the hydroxylation of phenylalanine and is a precursor for synthesis of proteins, thyroid hormones, melanin, and catecholamines (Mitchell et al., 2001). Excess tyrosine is either decarboxylated to tyramine by tyrosine decarboxylase, a minor pathway, or degraded to fumarate and acetoacetate by the sequential action of five enzymes: tyrosine aminotransferase catalyzes the transamination of tyrosine to p-hydroxyphenylpyruvate; p-hydroxyphenylpyruvate dioxygenase catalyzes the oxidation of p-hydroxyphenylpyruvate to homogentisate (HGA); homogentisate dioxygenase catalyzes the oxidation of HGA to maleylacetoacetate (MAA); glutathione transferase zeta/maleylacetoacetic acid isomerase (GSTZ1-1) catalyzes the isomerization of MAA to fumarylacetoacetate (FAA); and fumarylacetoacetate hydrolase catalyzes the hydrolysis of FAA to fumarate and acetoacetate (Knox and LeMay-Knox, 1951; Mitchell et al., 2001).
Deficiencies of tyrosine aminotransferase, p-hydroxyphenylpyruvate dioxygenase, and homogentisate dioxygenase are associated with benign inborn errors of metabolism (Mitchell et al., 2001). Loss-of-function mutations of fumarylacetoacetate hydrolase are, however, associated with the severe metabolic disorder hereditary tyrosinemia type 1 (HT-1). Acute HT-1 is characterized by complete fumarylacetoacetate hydrolase deficiency with fulminant liver failure and neonatal death (Kubo et al., 1998; Lindblad et al., 1977). Chronic HT-1 is characterized by partial loss of fumarylacetoacetate hydrolase activity with hepatocellular carcinomas, nephropathies (Fanconi’s syndrome), and neuropathies (Mitchell et al., 2001). The multiorgan damage is associated with the formation and accumulation of FAA and its derivatives, succinylacetoacetate (SAA) and succinylacetone (SA); the mechanism of formation of SAA and SA from FAA is poorly understood (Lindblad et al., 1977; Sassa and Kappas, 1983). Other biochemical changes in HT-1 are elevated serum and urine concentrations of tyrosine, p-hydroxyphenylpyruvic acid, methionine, and δ-aminolevulinic acid (δ-ALA) (Berger et al., 1983). The elevated excretion of δ-ALA in HT-1 has been associated with SA-induced inhibition of δ-aminolevulinic acid dehydratase (δ-ALAD) (Sassa and Kappas, 1983). HT-1 is diagnosed by determining SA concentrations in urine through GC-MS analysis (Lindblad and Steen, 1982; Lindblad et al., 1977; Schierbeek and Berger, 1989; Tuchman et al., 1984) or through the δ-ALAD inhibition assay (Grenier et al., 1982; Schulze et al., 2001).
Unlike other enzymes of the tyrosine degradation pathway, little is known about inborn metabolic errors associated with GSTZ1-1 deficiency. A genetic deficiency of GSTZ1-1, referred to as tyrosinemia type 1b, has been reported (Berger et al., 1988). Recent studies showed that GSTZ−/− mice are viable and excrete elevated amounts of FAA and SA, and that their viability is compromised when fed a high tyrosine-containing diet (Fernández-Cañón et al., 2002). FAA excretion in these mice was an unexpected finding, because FAA would be expected to be hydrolyzed by FAA hydrolase to fumarate and acetoacetate (Knox and Edwards, 1955) but indicates that GSTZ1-1 deficiency may cause biochemical changes similar to those seen in HT-1.
In addition to its activity with MAA, GSTZ1-1 also catalyzes the biotransformation of a range of xenobiotic α-haloalkanoic acids, including dichloroacetic acid (DCA) (Tong et al., 1998a,b). DCA is an investigational therapeutic agent that is used for the clinical management of lactic acidotic disorders, including congenital lactic acidosis, mitochondrial encephalopathy, lactic acidosis, and stroke-like syndrome (MELAS) (Saitoh et al., 1998). DCA is a metabolite of the sedative chloral hydrate (Stacpoole et al., 1998).
DCA is a mechanism-based inactivator of GSTZ1-1 (Anderson et al., 1999, 2002; Tzeng et al., 2000). DCA-inactivated GSTZ1-1 lacks maleylacetoacetate isomerase activity (Lantum et al., 2002a). GSTZ1-1 expression and activities are decreased below detection limits in hepatic and some extrahepatic tissues in DCA-treated rats, indicating that the DCA-modified GSTZ1-1 protein is rapidly degraded (Anderson et al., 1999; Lantum et al., 2002c). DCA treatment may, therefore, reduce flux through the tyrosine degradation pathway, but the consequences of this reduced flux are unclear.
The objective of this study was to determine the effects of DCA on the excretion of the MAA-derived intermediates MA, FA, and SA in rats. The cytotoxicity of MA, FA, and SA was also determined with a liver-derived cell line. The data presented in this paper show that DCA induced the excretion of MAA and MA in a manner that was dependent on flux through the tyrosine degradation pathway and that MA was a cytotoxic metabolite of tyrosine.
MATERIALS AND METHODS
Chemicals.
DCA (>99% pure), glutathione, HGA, δ-ALA, porphobilinogen, SA (4,6-dioxoheptanoic acid), hydroxylamine HCl, N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylsilyl chloride, p-dimethylaminobenzaldehyde (Ehrlich’s reagent), trichloroacetic acid, and mercuric chloride were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). [15N]-hydroxylamine HCl was purchased from ICN Stable Isotopes (Cambridge, MA). MA and FA were a gift from Dr. Peter Dedon, MIT, and were synthesized by the method of Fowler and Seltzer (1970).
Animals.
Male Fischer 344 rats (200–250 g; Taconic Farms, Germantown, NY) were housed individually in metabolism cages and were provided with standard rodent chow (Purina, St. Louis, MO) and double-distilled water ad libitum. The animal rooms were humidity- and temperature-controlled, and the rats were exposed to 12-h dark-light cycles. The protocol for animal use was reviewed and approved by the University Committee on Animal Resources. The rats were allowed 1 day to adapt to the metabolism cages before the start of each experiment.
Rats were given saline, DCA (0.3, 0.6, or 1.2 mmol/kg/day), and HGA (0.25 mmol/kg/day) ip as described in the figure legends. The solutions were prepared daily in 0.9% saline (Abbott Laboratories, North Chicago, IL) and were brought to pH 7.3 ± 0.2 by addition of NaOH. The solutions were filter-sterilized with 0.22-μm filter units (Sterivex-GV; Millipore, Bedford, MA). The doses of DCA used in this study have previously been shown to deplete GSTZ1-1 protein and activities in the liver and other organs of rats (Anderson et al., 1999; Lantum et al., 2002c). Moreover, 0.3, 0.6, and 1.2 mmol DCA/kg are similar to the doses used for the clinical management of congenital lactic acidosis (Stacpoole et al., 1998). In preliminary studies, doses of HGA above 0.25 mmol/kg failed to enhance the MAA excretion above that observed with 0.25 mmol/kg; hence, a dose of 0.25 mmol/kg was used. Finally, elevated amounts of HGA are deposited in tissues as dark brown pigments, which causes ochronosis (Mitchell et al., 2001).
Rats were divided into groups of three for each experiment and were injected with the study compounds dissolved in 1.5 ml saline once daily between 9.00 and 10.00 a.m. Urine samples, collected in plastic containers every 6–12 h, were centrifuged at 3,000 r.p.m. to remove food particles. The urine samples from individual rats were pooled as 24-h samples and were stored at −80°C until urinalysis.
Morphological analysis.
Animals were anesthetized with diethyl ether and sacrificed by decapitation 24 h after the last treatment. Sections of liver, brain, and kidney were excised and immediately immersion-fixed in 250 ml of 10% (w/w) phosphate-buffered formalin for 12 h, dehydrated with serial ethanol cycles, then embedded in paraffin. The paraffin-embedded tissue was cut into 5-μm sections and mounted on Superfrost plus slides (VWR Scientific Products, West Chester, PA). The tissue sections were deparaffinized and stained with Mayer hematoxylin and eosin stains for light microscopic analysis. The slides were also analyzed for in situ cell death by the direct transferase-mediated dUTP nick end labeling (TUNEL) fluorescence assay, following manufacturer instructions (Roche, Indianapolis, IN). The slides were coded before submission to the pathologist for analysis.
Gas chromatography-mass spectrometric (GC-MS) analysis of MA, FA, and SA in urine.
Methods to determine SA concentrations in urine developed by Tuchman et al. (1984) and Schierbeek and Berger (1989) were used with slight modifications. Briefly, to 1 ml of thawed urine was added 0.4 mmol (50 μl of 552 mg/ml) hydroxylamine HCl and 30 μl of concentrated HCl (final pH ~2.8). The samples were mixed and incubated at 80°C for 1 h in closed 2-ml Eppendorf tubes. After allowing the samples to cool, 1 μmol of [15N]-fumarylacetone oxime ([15N]-5(3)-methyl-3(5)-isoxazolepropenoic acid), prepared by incubating 15.6 mg FA with a 10-fold molar excess (70 mg) [15N]-hydroxylamine HCl in 5 ml acidified double-distilled water for 1 h at 80°C, was added to each sample as internal standard; 50 μl of 100 mM NaCl was also added to each sample, and the samples were mixed. The mixture was extracted twice with ethyl acetate (500 μl), and the pooled organic fractions were dried in a SpeedVac centrifuge (Savant Instruments, Holbrook, NY) at room temperature for 1 h. The dried residue was dissolved in 50 μl of pyridine, then derivatized by addition of 50 μl of MTBSTFA and heating at 80°C for 30 min. The derivatized samples (1 μl) were then analyzed on a Hewlett Packard 5720 GC/5972 MSD equipped with an HP-1 column (cross-linked methyl siloxane, 25 m long × 0.2 mm i.d., 0.5-μm film thickness, Hewlett Packard, Wilmington, DE) and a splitless liner. The temperatures of the injector port and detector were 230°C and 280°C, respectively; the initial oven temperature was held at 100°C for 3 min, increased to 275°C at 5°C/min, then held at 275°C for 5 min; the column pressure was set at 62 kPa; the carrier gas was 99.9% helium (0.5 ml/min). The tert-butyldimethylsilyl (tBDMS) derivatives of MA, SA, and FA had retention times of 29.2, 29.5, and 31.2 min, respectively. Scan and selected-ion monitoring (SIM) methods were used for quantification. The amounts of MA, SA, and FA present were determined from calibration curves ([peak area of analyte/peak area of internal standard] vs. amount of analyte) for MA, FA, and SA diluted in either saline or urine from normal rats. The calibration curves were linear for all analytes over the range of 0–1,000 μg/ml. The recovery of each analyte was determined by comparing peak areas of analytes dissolved in urine with those dissolved in saline prior to workup; the recovery was > 95 % for SA and 27% for MA. The lowest detectable amount was 6–10 nmol for MA and SA diluted in saline and 50–100 nmol for MA and SA diluted in urine. Approximately 45–75% of MA added to blank urine samples was recovered as FA, indicating isomerization of MA to FA during sample preparation (Fowler and Seltzer, 1970).
Liquid chromatography-mass spectrometric (LC-MS/MS) analysis of urine.
To 1 ml of each urine sample were added 50 μl concentrated HCl and 50 μg salicylic acid as internal standard. (Salicylic acid was chosen because it is not found in control rat urine and forms parent negative ions with m/z in the same range as those of MA, FA, and SA.) The urine samples were mixed and centrifuged at 13,000 r.p.m. for 15 min, and 50 μl samples were analyzed by reverse-phase electrospray ionization LC-MS/MS on an Agilent 1100 Series MSD Trap (Agilent, Palo Alto, CA) in the negative-ion mode. The liquid chromatograph was equipped with an Eclipse XDB-C8 column (5 μm particle size, 4.6 μm × 150 mm, Agilent), which was eluted with a 0–95% gradient at a flow rate of 0.3 ml/min over 20 min; solvent A contained 2 mM ammonium acetate (pH 6.8) dissolved in Omnipur water (EM Science, Merck KGa, Darmstadt, Germany), and solvent B contained 2 mM ammonium acetate in HPLC-grade acetonitrile (J.T Baker, Phillipsburg, NJ). The retention time was 10–11 min for MA, 12 min for FA, and 14 min for SA. The MS/MS data were acquired in auto and manual mode at fragmentation voltages of 0.35 and 0.65 V. The ionization and fragmentation parameters were optimized to obtain parent ions of SA (157 m/z), and MA and FA (155 m/z) by direct infusion of a 400 μM solution of SA, MA, or FA dissolved in a solution containing 50% of solvents A and B. Diagnostic fragment ions for MA and FA were observed at 155 ([M-H]−), 137, and 111 m/z; and diagnostic ions for SA were observed at 157 ([M-H]−), 139, and 113 m/z. The recoveries of MA, FA, SA, and salicylic acid (1–100 μg) diluted in urine, compared with the same amounts diluted in saline, were 15–17%, indicating reaction or adsorption of the analytes to matrix components under experimental conditions or ion suppression at the detector by coeluting compounds. Compared with the GC-MS method, the LC-MS/MS method afforded shorter analysis times and easier sample preparation. LC-MS/MS analyses of urine samples were, therefore, used for qualitative and semiquantitative analysis of analytes.
Enzymatic synthesis of MAA.
MAA was synthesized enzymatically by the method of Knox and Edwards (1955). Briefly, 5 mM HGA was incubated with 10 mg of dialyzed rat liver homogenate in 200 mM phosphate buffer (pH 7.0) containing 2 mM ascorbic acid and 50 μM FeSO4. The reaction mixture was incubated for 1 h at 37°C, acidified with 50 μl of concentrated HCl, extracted with ethyl acetate after addition of NaCl, and derivatized to form the tBDMS derivative, as described above. A 50-μl sample of the reaction mixture was also analyzed by LC-MS/MS after acidification and removal of precipitated proteins by centrifugation.
δ-ALAD activities.
δ-ALAD activities were determined by the method of Mauzerall and Granick (1956), adapted for test-tube-size experiments and for 96-well plate experiments (Anderson and Desnick, 1979; Schulze et al., 2001). Crystalline δ-ALAD (5 U/3.8 mg dry weight; Sigma-Aldrich) was dissolved in 500 μl of 100 mM Tris–HCl buffer (pH 6.8) containing 6 mM DTT or 1 mM TCEP (tris[2-carboxyethyl]phosphine hydrochloride, Pierce Biotechnology, Inc., Rockford, IL) and incubated for 15 min at 37°C to activate the enzyme immediately before use.
For urine δ-ALAD inhibition assays, 100 μl of urine from saline-treated rats or from rats given 1.2 mmol/kg/day DCA or 0.25 mmol/kg/day HGA, or both, was added to reaction mixtures containing 4 mM δ-ALA, 2 mM TCEP, 0.5 U of activated enzyme, and 400 μl 100 mM Tris–HCl buffer (pH 6.8). After incubation for 30–60 min at 37°C, the reaction was quenched by addition of 250 μl of stop reagent (0.05 M mercuric chloride in 10% trichloroacetic acid). The colorimetric reaction was started by addition of 750 μl of modified Ehrlich’s reagent, prepared immediately before use by mixing 8 ml of 70% perchloric acid and 1 g of p-dimethylaminobenzaldehyde in 42 ml of glacial acetic acid (Mauzerall and Granick, 1956), to the reaction mixture. After standing for 15 min at room temperature, the absorbance of the solutions was measured at 555 nm against water.
MA-, FA-, and SA-inhibition assays were conducted in 96-well plates. The reaction mixtures contained 0–2.5 mM δ-ALA in 50 μl of 100 mM Tris–HCl (pH 6.8), without or with inhibitors, and were incubated for 15 min at 37°C. The reactions were started by addition of 10 μl (~ 16 mU) of activated enzyme to each well and incubated for 1.0 h at 37°C with 10-sec shake-cycles every 15 min. The reaction was stopped by addition of 50 μl of stop reagent and 100 μl of modified Ehrlich’s reagent. The absorbance was measured at 550 nm with an MR5000 microplate reader (Dynatech Laboratories Inc., Chantilly, VA) after standing for 15 min at room temperature. The amount of porphobilinogen formed was determined from a calibration curve prepared with known amounts of porphobilinogen (0–20 nmol) diluted in 100 mM Tris–HCl. The competitive inhibition constants were determined by Dixon plots (1/x vs. [I]) of data obtained from each experiment and averaged for three experiments.
Cell culture.
TGF-α Transgenic mouse hepatocytes (TAMH) were chosen for the viability studies because they are readily dispersed as single cells suitable for flow cytometry with minimal trituration; TAMH cells, although transformed, express drug-metabolizing enzymes and differentiated hepatocyte-markers in a passage-independent manner and do not require serum and growth factors for growth or viability, unlike primary hepatocytes and many other hepatocyte-derived cell lines (Pierce et al., 2002). TAMH cells were cultured in Dubelcco’s modified Eagle’s medium (Gibco), supplemented with 5 μg/ml insulin, 5 μg/ml transferrin, 5 ng/ml selenium (Collaborative Biomedical Products), and 0.1 μmol/ml dexamethasone, 50 μg/ml gentamicin, and 10 mM nicotinamide. The cells were grown in flat-bottomed 35-mm diameter 6-well dishes (Corning, NY) in a humidified incubator with 5% carbon dioxide/95% atmospheric air and passaged once weekly.
Cell viability assays.
Cell viability was determined with the Live/Dead® Cytotoxicity Assay Kit (Molecular probes, Eugene, OR) according to the manufacturer’s instructions; the assay is based on the intracellular esterase-catalyzed cleavage of nonfluorescent cell-permeant calcein-acetoxymethyl ester (calcein AM) to the green-fluorescent calcein in live cells and the nuclear staining by cell-impermeant red-fluorescent ethidium homodimer in dead cells. Briefly, cells were grown to ~80% confluence and incubated with 0–1 mM MA or FA dissolved in 0.05 M phosphate buffer (pH 7.2). After a 6-h incubation, the medium was transferred to 10-ml polystyrene tubes to recover free cells, and the adherent monolayer of cells was detached by treatment with 0.25% trypsin-1 mM EDTA solution (GibcoBRL, Life Technologies Inc., Grand Island, NY) and added to the suspension of recovered free cells. The pooled cell suspensions were diluted 5-fold with ice-cold PBS and washed twice by repeated trituration and centrifugation. The final cell pellet was suspended in 100 μl PBS and 100 μl of Live/Dead® assay reagent containing 2 μM calcein AM and 4 μM ethidium homodimer, and kept at room temperature for 5 min before analysis by flow cytometry (FACSCalibur®, Becton, Dickinson, and Company, Silicon Valley, San Jose, CA) with excitation at 488 nm and emission at 530 nm for the green-fluorescent signal and at 585 nm for the red-fluorescent signal. For each cell suspension, 25–50,000 events within a previously established gate defined by forward- and side-scatter properties of untreated cells were scored, and bivariate frequency distributions were used to determine the percentage of live cells in the total cell count.
Statistical analyses.
The data were analyzed by two-way ANOVA with Bonferroni’s multiple comparison test (GraphPad Prism®, GraphPad Software Inc., San Diego, CA). A level of p < 0.05 was chosen for acceptance or rejection of the null hypothesis. The kinetic data were analyzed by nonlinear regression analysis (GraphPad Prism®) or fitted to the Michaelis-Menten equation with EnzFitter software (Biosoft, Ferguson, MO).
RESULTS
Inhibition of δ-ALAD Activity
Urine from rats given 0.25 mmol/kg/day HGA or 1.2 mmol/kg/day DCA, or both, were tested for inhibition of δ-ALAD activity. Urine from HGA-treated rats was used as a control to avoid the effects of administered HGA on the excretion of MAA-derived metabolites in the absence of DCA treatment. (The activities of δ-ALAD in the presence of urine from rats given HGA for 5 days were less than the activities determined in the presence of urine collected from rats given saline for 5 days. The reason for the lower activity in the presence of urine from HGA-treated rats compared with saline-treated rats is not known.)
Urine from rats given DCA or both DCA and HGA for 5 days markedly inhibited δ-ALAD activity, compared with urine from rats given HGA (Fig. 1), indicating that rats given DCA excreted a compound that inhibited δ-ALAD. These data are similar to the δ-ALAD-inhibiting effects of urine from HT-1 patients and are consistent with the presence of SA in the urine (Lindblad et al., 1977).
Inhibition of δ-ALAD by MA, FA, and SA
Because of the structural similarities between SA and both MA and FA, we hypothesized that MA or FA, or both, may inhibit δ-ALAD. Hence, the inhibitory effects of MA, FA, and SA on δ-ALAD were determined by steady-state inhibition kinetic analysis of δ-ALAD activity. All three compounds were competitive inhibitors of δ-ALAD (Fig. 2); SA was the most potent inhibitor with a Kic of 0.15 ± 0.05 μM, followed by FA (Kic ~ 4 ± 1 μM) and MA (Kic ~ 20 ± 3 μM). These data indicated that the δ-ALAD inhibition assay was not specific for MA, FA, or SA (Sassa and Kappas, 1983).
Mass Spectrometric Analysis of Urine of Rats Given DCA
Analysis of urine from HT-1 patients by GC-MS shows elevated SA excretion (Lindblad et al., 1977; Tuchman et al., 1984), and the base peak in the mass spectrum of the tBDMS derivative of SA is seen at m/z 212 (Schierbeek and Berger, 1989). Hence, the dose- and time-dependence of the area-under-the-curve for the abundance of m/z 212 ions in SIM mode in the urine of rats given 0, 0.3, 0.6, and 1.2 mmol DCA/kg body weight was determined.
DCA treatment resulted in a dose- and time-dependent increase in the abundance of the m/z 212 ions in the urine. The abundance of the m/z 212 ions after giving the first dose of DCA was not different from control. Beyond the second day of treatment, however, the abundance of the m/z 212 ions increased significantly above control values in rats given 0.6 and 1.2 mmol DCA/kg/day; the abundance of the m/z 212 ion was maximal after three days of treatment for rats given 1.2 mmol/kg and at days 4 and 5 in rats given 0.3 and 0.6 mmol/kg, respectively (Fig. 3). The reason for the decline in the abundance of the m/z 212 ion beyond the third day in rats given 1.2 mmol/kg DCA is not known.
The identity of the m/z 212 ions could not be unequivocally established by GC-MS in the SIM mode because the tBDMS derivatives of MA and SA had similar retention times (tRMA ~ 29.2 min; tRSA ~ 29.5 min). The m/z 212 ion could be the base peak for tBDMS-SA, which is consistent with its published spectrum (Schierbeek and Berger, 1989), or the m/z 212 ion could be the “M + 2” isotopic peak, where M is the m/z 210 base peak of tBDMS-MA (Fig. 4). (tBDMS-MA and tBDMS-FA had identical spectra with a base peak at m/z 210 but had different retention times (tRMA ~ 29.2 min vs. tRFA ~ 31.2 min, data not shown). Subsequent GC-MS experiments were done in scan mode, and peak areas for the extracted-ion chromatograms of m/z 210 and 212 were used for quantification of the tBDMS derivatives of MA and FA, as well as SA, respectively.
GS-MS analysis (scan mode) of the urine from rats given 1.2 mmol/kg/day DCA showed the presence of tBDMS-MA. tBDMS-FA was observed in the urine of DCA-treated rats and was derived from the spontaneous isomerization of MA to FA during sample workup. The excretion of MA increased with days of treatment and was enhanced by the coadministration of HGA (Fig. 5). MA excretion was confirmed by LC-MS/MS analysis of the urine (data not shown). SA was not detected by GC-MS and LC-MS/MS analyses of the urine of rats given DCA, HGA, or both.
Another compound with a m/z 210 ion was observed in some urine samples of rats given both DCA and HGA (Fig. 6A); GC-MS analysis showed that this compound had a similar retention time (tR = 35.1 min) to that of enzymatically synthesized MAA. The mass spectral characteristics of MAA were confirmed by LC-MS/MS analysis (Fig. 6B). MAA excretion was not quantified but appeared to increase with days of treatment with DCA (data not shown). DCA treatment, therefore, induced the excretion of MAA and MA. It is uncertain whether MAA undergoes decarboxylation to MA in the tissues or in the urine.
Reversibility of DCA-Induced Excretion of MA
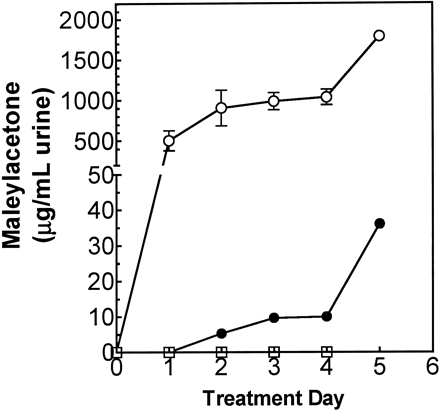
The recovery from the effects of DCA treatment on MA excretion was determined in rats given 1.2 mmol/kg DCA for 4 days, then 0.25 mmol/kg HGA for 3 subsequent days. As indicated above, DCA treatment increased the excretion of MA, and MA excretion was further increased by the first dose of HGA. MA concentrations returned to near baseline values within 48 h after the last dose of DCA, even after HGA treatment (Fig. 7). These data indicated that the excretion of MA was reversible after discontinuing DCA treatment and was not sustained by giving HGA alone. This observation is consistent with previous studies in our laboratory, which showed that hepatic GSTZ1-1 activities and protein levels return to normal levels within 48 h after the last dose of DCA (Anderson et al., 1999).
Rats given equimolar doses of chlorofluoroacetic acid, which is a noninactivating analogue of DCA (Anderson et al., 1999), failed to excrete MA, even after coadministration of HGA. These data indicate that the DCA-induced inactivation of GSTZ1-1 leads to the increased excretion of MA.
Cytotoxicity of MA and FA
The cytotoxicity of MA was determined in TAMH cells, as described in Materials and Methods. MA caused a decrease in viability of TAMH cells, with a steep dose-response curve; the EC50 of MA was ~350 μM after a 6-h incubation; MA was less potent than FA (EC50 ~250 μM) (Fig. 8A). (GSTZ1 expression was undetectable in homogenates of TAMH cells by Western blot analysis with polyclonal anti-GSTZ1 antibodies [Board et al., 1997; Lantum et al., 2002c]). Nonenzymatically formed FA may, therefore, contribute to the observed MA-induced cytotoxicity.
Because of the structural similarity of MA and FA to medium-chain fatty acids, we hypothesized that MA and FA may undergo β-oxidation-dependent bioactivation to reactive intermediates. Octanoic acid and benzoic acid inhibit β-oxidation-dependent bioactivation pathways (Fitzsimmons and Anders, 1993). Hence, the effects of octanoic acid and benzoic acid on the toxicities of MA and FA with TAMH cells were determined. Octanoic acid and benzoic acid did not alter the toxicity of MA and FA, indicating the lack of a role for β-oxidation-dependent bioactivation (Fig. 8B).
Morphological Examination of Tissues from Rats Given DCA Alone or with HGA
Because MA was cytotoxic in vitro, sections of liver, kidney, and brain tissue from rats given 0.25 mmol/kg HGA, 1.2 mmol/kg DCA, or both, for 5 days were analyzed for cytotoxicity by light microscopy. (Hepatic, renal, and central nervous system toxicities are observed in HT-1 patients [Anderson et al., 2002; Mitchell et al., 2001]). Significant tissue damage was not observed in any of the tissues in all treatment groups (data not shown). The absence of toxicity was confirmed by TUNEL-staining and fluorescence microscopy of the tissue sections, and by determining caspase activation through Western blot analysis of tissue homogenates of the same rats (data not shown).
DISCUSSION
DCA is a mechanism-based inactivator of GSTZ1-1 (Tzeng et al., 2000). The DCA-induced inactivation of hGSTZ1-1 results in lowered maleylacetoacetate isomerase activities of recombinant hGSTZ1-1 (Lantum et al., 2002a) and lowered GSTZ1-1 activities in most rat tissues (Anderson et al., 1999; Lantum et al., 2002c). Long-term treatment with DCA is associated with multiorgan toxicity in rodents and dogs (Stacpoole et al., 1998) that is similar to the multiorgan toxicities seen in HT-1. DCA toxicity in humans is heralded by ataxia after 6 weeks of treatment (Stacpoole et al., 1998). The data presented in this study describe some of the biochemical changes that are associated with perturbation of MAA-metabolism in rats given DCA for 5 days.
MA Excretion in DCA-Treated Rats
GC-MS and LC-MS/MS methods were developed to characterize the metabolites excreted after treatment of rats with DCA. DCA-treated rats excreted elevated amounts of MA, as reported previously by Cornett et al.(1999). FA was also observed in the urine samples of DCA-treated rats. Because of the known isomerization of MA to FA that occurs in aqueous solutions (Fowler and Seltzer, 1970), it is likely that the FA present in urine is formed by the isomerization of MA before or during sample preparation. In addition to MA excretion, MAA excretion was also observed in the urine of some rats given both DCA and HGA. The MA present in urine may arise from the decarboxylation of MAA (Fowler and Seltzer, 1970; Knox and Edwards, 1955).
The mean daily excretion of MA beyond the third day of DCA treatment ranged between 100 and 500 μg/24 h (0.6–3.0 μmol/24 h). MA excretion was enhanced tenfold by the coadministration of HGA. The excretion of MA returned to baseline within 48 h after the last dose of DCA, even in the presence of HGA. These data indicated that the excretion of MA in DCA-treated rats was determined by the flux through the tyrosine degradation pathway and was reversible. Continuous administration of DCA is, therefore, required to sustain MA excretion. These findings differ from those observed in GSTZ−/− mice fed a standard diet, which show an elevated excretion of FAA and SA (Fernández-Cañón et al., 2002). The reason for this difference is not immediately apparent but is unlikely to be associated with species differences, given that the enzymes of the tyrosine degradation pathway, especially GSTZ1-1, are highly phylogenetically conserved (Board et al., 1997; Mitchell et al., 2001). It is possible that the FAA observed in the urine of GSTZ−/− mice arises from the isomerization of MAA. (We have not observed the spontaneous reduction of MA or FA to SA in vitro [H. B. M. Lantum and M. W. Anders, 2002, unpublished observations]).
Lack of SA Excretion in DCA-Treated Rats
SA was not detected in the urine of rats given DCA, indicating that MAA or MA were not reduced to SAA or SA in vivo, although previous studies indicated that MAA or MA are reduced to SAA or SA (Fernández-Cañón et al., 2002) (Figure 9). The observation that SA was not detected in the urine of DCA-treated rats parallels the results obtained from a patient with tyrosinemia type Ib (due to a deficiency of GSTZ1-1), who also failed to excrete SA (Berger et al., 1988). Hence, the lack of SA excretion in rats with lowered GSTZ1-1 activities indicates that SA does not underlie the pathophysiology of DCA-induced toxicities and tumors.
SA-induced inhibition of DNA ligase-1 and S-adenosyl methionine synthetase are implicated in perturbation of DNA homeostasis and tumorigenesis in HT-1 (Berger et al., 1983; Prieto-Alamo and Laval, 1998). Perturbation of DNA homeostasis, marked by increased DNA single-strand breaks and DNA hypomethylation, is also observed in DCA-treated rats (Tao et al., 2000). The role of MA and FA in DCA-induced perturbation of DNA homeostasis merits investigation.
Inhibition of δ-ALAD by MA and FA
DCA-treated rats excreted metabolites that inhibited δ-ALAD; the urine from patients with HT-1 also inhibits δ-ALAD (Lindblad et al., 1977). Nephrotoxicity and neurotoxicity in HT-1 are associated with SA-induced inhibition of δ-ALAD and a buildup of δ-ALA (Wyss et al., 1993). The finding that DCA-treated rats excreted MA and FA but not SA indicated that other compounds that inhibit δ-ALAD are excreted in the urine of DCA-treated rats. Steady-state inhibition kinetic analyses showed that MA and FA competitively inhibited δ-ALAD but were less potent than SA. This is the first report of MA- and FA-induced inhibition of DTT- or TCEP-activated δ-ALAD. Berger et al.(1983) described noncompetitive inhibition of δ-ALAD by FAA in the absence of DTT, but DTT was required for activity in our experiments. Inhibition of δ-ALAD by MA or FA, or both, may contribute to the porphyria observed in HT-1 patients, which is marked by neurological crisis (Gentz et al., 1969; Kappas et al., 2001). Similarly, inhibition of δ-ALAD by 4,6-dioxoheptenoic acid metabolites formed in DCA-treated rats may also perturb heme biosynthesis in vivo and lead to porphyria-like symptoms. Decreased erythrocyte counts and hemoglobin concentrations are observed in dogs after 30 days of treatment with DCA (Cicmanec et al., 1991).
An assay based on inhibition of δ-ALAD is used for neonatal screening for HT-1 (Schulze et al., 2001). Because MA and FA also inhibited δ-ALAD, the specificity of the δ-ALAD inhibition assay for diagnosing the excretion of SA merits examination. The LC-MS/MS method and GC-MS scan method described in this study may be better-suited alternatives for identifying metabolites of the tyrosine degradation pathway associated with fumarylacetoacetate hydrolase and GSTZ1-1 deficiencies, compared with the δ-ALAD inhibition assay.
Disposition and Toxicity of MA and FA
Tissue concentrations of MA and FA were not measured in the present studies. It has, however, been reported that SA accumulates in kidney and brain tissue (~0.8 μmol/g protein) more than in liver tissue (~0.2 μmol/g protein) after ip administration of SA to rats (Wyss et al., 1993) and that these tissue concentrations of SA are associated with perturbation of heme homeostasis (Wyss et al., 1992). The data presented herein may indicate that tissue concentrations of MA sufficient to perturb heme homeostasis are unlikely to be attained in rats that are fed a standard diet and given DCA alone.
Cytotoxicity studies showed that MA was cytotoxic at concentrations greater than 150 μM. The cytotoxicity of MA was not blocked by benzoic or octanoic acid, indicating the lack of a role for β-oxidation-dependent bioactivation of MA. MA and FA are alkylating compounds that covalently modify thiol moieties in proteins by Michael-addition reactions (Lantum et al., 2002b). Cellular targets that undergo alkylation by MA and FA have not been identified, but unpublished data indicate that MA and FA are potent inactivators of recombinant P-glycoprotein and poor inhibitors of recombinant caspase-3, both of which are cysteine-dependent enzymes, indicating selectivity in the alkylating effects of MA (H. B. M. Lantum and M. W. Anders, 2002, unpublished observations).
Despite the elevated excretion of MA in rats given DCA alone or with HGA, morphological changes were not observed by light microscopic analysis of liver, kidney, and brain tissues after 5 days of treatment. These findings are consistent with observations made in GSTZ−/− mice, which did not show apparent signs of toxicity when fed a standard rodent diet but developed liver and renal toxicities and died within 5–50 days when given HGA or when fed phenylalanine-, tyrosine-, or protein-rich diets (Fernández-Cañón et al., 2002). The lack of acute toxicity in DCA-treated rats in the present study may be due to low flux through the tyrosine degradation pathway under physiological conditions, the short duration of the treatment, the lack of a buildup of MA in tissues, or the rapid clearance of MA from tissues. Studies with prolonged administration of DCA in rats fed high tyrosine-containing diets may be instructive in understanding the pathogenesis of MA-associated toxicity.
The data presented in this study show that DCA-induced inactivation of GSTZ1-1 results in the excretion of MA, which, if present at concentrations greater than 150 μM in tissues, may cause cytotoxicity. These findings have important implications for understanding the pathophysiology of DCA-induced toxicities in humans and rodents and hepatocarcinogenesis in rodents (Stacpoole et al., 1998). The concentrations of DCA used in this study are much higher than those present in municipal water supplies, where DCA is formed as a by-product of the chlorination of water (Uden and Miller, 1983). DCA can be formed by the metabolism of trichloroethylene (Merdink et al., 1998), which is an industrial solvent, or by the metabolism of chloral hydrate, which is a clinically used sedative (Stacpoole et al., 1998). The use of DCA as an investigational therapeutic agent for clinical management of lactic acidotic disorders (Stacpoole et al., 1998) exposes humans to doses (~50 mg/kg/day) similar to those used in this study. The excretion of MA can, therefore, be used to search for metabolic changes associated with DCA therapy. The data presented herein and the results obtained with GSTZ–/–mice (Fernández-Cañón et al., 2002) may indicate that a low tyrosine- or protein-containing diet may limit flux through the tyrosine-degradation pathway; such dietary intervention merits investigation for its possible beneficial effects in subjects undergoing long-term DCA therapy.
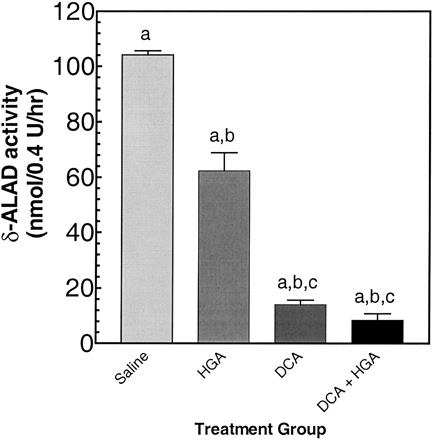
Inhibition of δ-ALAD by samples of urine from rats given DCA or both DCA and HGA. The activity of δ-ALAD was determined in urine samples from rats given 0.25 mmol/kg/day HGA (HGA), 1.2 mmol/kg/day DCA (DCA), or both (DCA+HGA), for 5 days, as described in Materials and Methods. Urine samples from rats given saline for 5 days are also shown. The data are presented as means ± SEM; n = 3 per group. Two-way ANOVA: (a) significantly different from Saline (p < 0.05); (b) significantly different from HGA (p < 0.05); (c) DCA compared with DCA + HGA, not significantly different (p > 0.05).
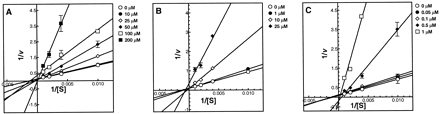
Lineweaver-Burk plots showing competitive inhibition of δ-ALAD by (A) MA, (B) FA, and (C) SA. MA, FA, and SA were incubated with δ-ALAD, and the amount of porphobilinogen formed was determined, as described in Materials and Methods. The data are presented as means ± SEM; n = 3.
![Dose-dependent effect of DCA on the excretion of 4,6-dioxohepta(e)noic acids. Rats were given saline or 0.3, 0.6, and 1.2 mmol/kg/day DCA for 5 days, and 24-h urine samples were analyzed by GC-MS in selected-ion monitoring mode for the presence of m/z 212 ions, as described in Materials and Methods. The m/z 212 ions may represent the base peak of the tBDMS derivative of SA or may be the “M + 2” peak, with M = 210 m/z, the base peak of tBDMS derivatives of MA and FA. The internal standard was the tBDMS-derivative of [15N]-succinylacetone oxime. Because analysis in selectedion monitoring mode does not distinguish between tBDMS-MA and tBDMS-SA, the data are presented in arbitrary units for each treatment group. The data are presented as means ± SEM; n = 3. *Two-way ANOVA: p < 0.05 for the group of DCA-treated rats compared with control on the specified treatment day.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/74/1/10.1093/toxsci/kfg104/2/m_120731350003.gif?Expires=1716464839&Signature=OAv81iyE9Eg7jl9jIQYz~lTuz9D~hGH3t5bVYBjPg1J4AoONRskq1dnju3bwxZCZXHDZNXYOFV6K7xINMZxUvrzIq8VNbyZkpVmu7BSttUxvF43FX47doU85JHws8D05lgPUyMEdVJNP3vSdI~SrHS7CalIvTH4nJEaaJmROdPku1axQyLxuXuchT3EL3jVvlL7tMEBgFTZewbkl5gwZs~vCppP6z1RB7RLbBKWKJ38KTnTCW~VOQ3wsObGMbItqA1uKrYUS7QY0bWUa92IaBYl3jjkfZbc8dOq~TCB2PXH0TFb5njfeJ58B7QF4bPpaYB2UyBeTI1sUAwumvr9iEw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Dose-dependent effect of DCA on the excretion of 4,6-dioxohepta(e)noic acids. Rats were given saline or 0.3, 0.6, and 1.2 mmol/kg/day DCA for 5 days, and 24-h urine samples were analyzed by GC-MS in selected-ion monitoring mode for the presence of m/z 212 ions, as described in Materials and Methods. The m/z 212 ions may represent the base peak of the tBDMS derivative of SA or may be the “M + 2” peak, with M = 210 m/z, the base peak of tBDMS derivatives of MA and FA. The internal standard was the tBDMS-derivative of [15N]-succinylacetone oxime. Because analysis in selectedion monitoring mode does not distinguish between tBDMS-MA and tBDMS-SA, the data are presented in arbitrary units for each treatment group. The data are presented as means ± SEM; n = 3. *Two-way ANOVA: p < 0.05 for the group of DCA-treated rats compared with control on the specified treatment day.
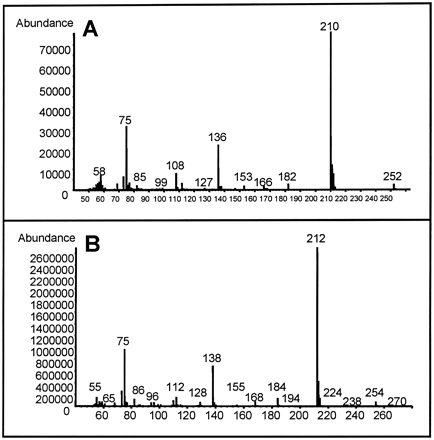
Mass spectra from urine to which 100 μg MA (A) or 100 μg SA (B) was added. MA- and SA-spiked urine samples were prepared for GC-MS analysis, as described in Materials and Methods. The mass spectrum for tBDMS-FA is identical with that of tBDMS-MA (data not shown). Urine samples from rats given DCA alone or with HGA contained compounds with a mass spectrum identical to that of tBDMS-MA (A) and had retention times consistent with those of tBDMS-MA and tBDMS-FA.
Effect of DCA treatment on MA and FA excretion in rats. Rats were given either 0.25 mmol HGA/kg/day (open square), 1.2 mmol DCA/kg/day (filled circle), or both (open circle), for 5 days, and 24-h urine samples were analyzed for the tBDMS-derivative of MA and FA by GC-MS in scan mode, as described in Materials and Methods. The amounts of MA and FA determined were summed, and the data are presented as means ± SEM; n = 3 per group. Two-way ANOVA: DCA and DCA + HGA groups had significantly (p < 0.05) elevated MA concentration, compared with control on all treatment days, except day 1 for DCA-treated rats.
![MAA excretion in rats given DCA. (A) Mass spectrum of di-tBDMS derivative of MAA obtained by GC-MS analysis of urine of rats given 1.2 mmol/kg/day DCA and 0.25 mmol/kg HGA for 5 days. (B) Extracted-ion chromatogram (EIC, top panel) and mass spectrum of MA ([M-H]− = 155 m/z; left bottom panel) and MAA ([M-H]− = 199 m/z; right bottom panel) obtained by LC-MS/MS (negative-ion mode) analysis of urine from rats given 1.2 mmol/kg/day DCA and 0.25 mmol/kg HGA for 5 days. The GC-MS and LC-MS/MS spectra of MAA were identical with that of enzymatically synthesized MAA.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/74/1/10.1093/toxsci/kfg104/2/m_120731350006.gif?Expires=1716464839&Signature=kkVxPgjuIwbxvngF~WhRvR1QG8oOoICt4HzTrlMmrPR7vwa~Ey48lJlOlTTWd~rwc70RfJtIVod9hO103DiHjpfNXYR~RhSUUgkIYNl6TkPcEGlReEwcrjm2UfihgFV9TzHQxmBFDIZkqSErWEJ1PyI2geLwY6OKyDv0Db8mV-bjcVLEJJqN8HfDayN8dfgJRUMRSpucqPAZ3re-OrIRnk1T7OUqVpK-IsG0obMU5gGEMylb14ustuGm2We4GdlnL6D2rruj2r3BgAxyff4bxkKfd9y7lXsX~x3h7A8LlB7b5DAb5zobq9TqM-0sqC183CCJ3rpaOfGGp5VFnfEoYw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MAA excretion in rats given DCA. (A) Mass spectrum of di-tBDMS derivative of MAA obtained by GC-MS analysis of urine of rats given 1.2 mmol/kg/day DCA and 0.25 mmol/kg HGA for 5 days. (B) Extracted-ion chromatogram (EIC, top panel) and mass spectrum of MA ([M-H]− = 155 m/z; left bottom panel) and MAA ([M-H]− = 199 m/z; right bottom panel) obtained by LC-MS/MS (negative-ion mode) analysis of urine from rats given 1.2 mmol/kg/day DCA and 0.25 mmol/kg HGA for 5 days. The GC-MS and LC-MS/MS spectra of MAA were identical with that of enzymatically synthesized MAA.
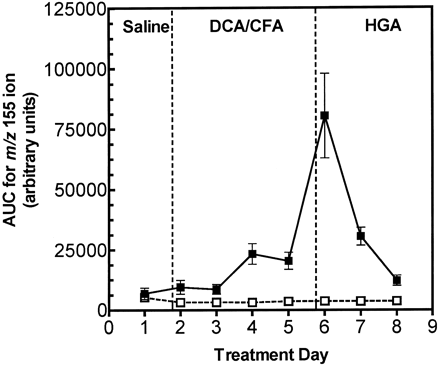
Reversibility of DCA-induced excretion of MA and FA, as measured by the abundance of m/z 155 ions in urine. Rats were given saline on day 1 prior to treatment with 1.2 mmol DCA/kg/day ip (filled square) or 1.2 mmol chlorofluoroacetic acid (CFA)/kg/day ip (open square) on days 2–5, then 0.25 mmol/kg/day HGA ip on days 6–8. The 24-h urine samples were analyzed by LC-MS/MS, and the area-under-the-curve (AUC) for MA was averaged for each sample and normalized with the AUC for the internal standard. The data are presented as means ± SEM; n = 3 rats per group. Two-way ANOVA: MA and FA excretion was significantly higher in DCA-treated rats, compared with CFA-treated rats on all treatment days beyond day 2.
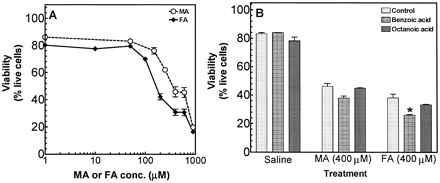
Cytotoxicity of MA and FA in TAMH hepatocytes. (A) Concentration-dependent cytotoxicity of MA and FA in TAMH hepatocytes. TAMH hepatocytes were incubated with 0–0.9 mM MA and FA for 6 h, and viability was determined by flow-cytometric analysis of cells loaded with calcein AM and ethidium homodimer, as described in the Materials and Methods. (B) Effects of benzoic acid and octanoic acid on the cytotoxicity of MA and FA. TAMH cells were incubated with 2 mM benzoic acid or octanoic acid, which was added to the culture medium 15 min prior to addition of 400 μM MA or FA for 6 h; viability was determined as described in Materials and Methods. Data are presented as means ± SEM; n = 3 replicates. *p < 0.05 for viability of cells incubated with FA and benzoic acid, compared with cells incubated with FA alone.
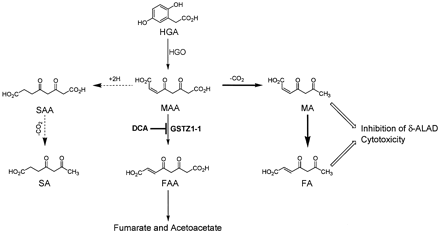
Pathway illustrating effects of DCA treatment on tyrosine degradation. Thin, full arrows represent the normal pathway of MAA metabolism; bold, full arrows represent pathways of MAA metabolism associated with DCA-induced GSTZ1-1 deficiency; broken arrows represent pathways of MAA metabolism that may not occur in DCA-induced GSTZ1-1 deficiency; open arrows represent effects of MA and FA.
Present address: Biochemical and Analytical Toxicology, Health Safety and Environmental Laboratories, Eastman Kodak Co., Rochester, NY 14652
To whom correspondence should be addressed at Department of Pharmacology and Physiology, University of Rochester Medical Center, 601 Elmwood Avenue, Box 711, Rochester, NY 14642. Fax: (585) 273-2652. E-mail: mw_anders@urmc.rochester.edu.
This research was supported in part by the University of Rochester Wilmot Cancer Research Fellowship program (H.B.M.L) and by the National Institutes of Environmental Health Sciences grants ES03127 and ES01247 (M.W.A.).
REFERENCES
Anderson, P. M., and Desnick, R. J. (
Anderson, W. B., Board, P. G., Gargano, B., and Anders, M. W. (
Anderson, W. B., Liebler, D. C., Board, P. G., and Anders, M. W. (
Berger, R., Michals, K., Galbraeth, J., and Matalon, R. (
Berger, R., van Faassen, H., and Smith, G. P. (
Board, P. G., Baker, R. T., Chelvanayagam, G., and Jermiin, L. S. (
Cicmanec, J. L., Condie, L. W., Olson, G. R., and Wang, S. R. (
Cornett, R., James, M. O., Henderson, G. N., Cheung, J., Shroads, A. L., and Stacpoole, P. W. (
Fernández-Cañón, J. M., Baetscher, M. W., Finegold, M., Burlingame, T., Gibson, K. M., and Grompe, M. (
Fitzsimmons, M. E., and Anders, M. W. (
Fowler, J., and Seltzer, S. (
Gentz, J., Johansson, S., Lindblad, B., Lindstedt, S., and Zetterstrom, R. (
Grenier, A., Lescault, A., Laberge, C., Gagné, R., and Mamer, O. (
Kappas, A., Sassa, S., Galbraith, R. A., and Nordmann, Y. (
Knox, E. W., and Edwards, S. W. (
Knox, W. E., and LeMay-Knox, M. (
Kubo, S., Sun, M., Miyahara, M., Umeyama, K., Urakami, K.-I., Yamamoto, T., Jakobs, C., Matsuda, I., and Endo, F. (
Lantum, H. B., Board, P. G., and Anders, M. W. (
Lantum, H. B., Liebler, D. C., Board, P. G., and Anders, M. W. (
Lantum, H. B. M., Baggs, R. B., Krenitsky, D. M., Board, P. G., and Anders, M. W. (
Lindblad, B., Lindstedt, S., and Steen, G. (
Lindblad, B., and Steen, G. (
Mauzerall, D., and Granick, S. (
Merdink, J. L., Gonzalez-Leon, A., Bull, R. J., and Schultz, I. R. (
Mitchell, G. A., Grompe, M., Lambert, M., and Tanquay, R. M. (
Pierce, R. H., Franklin, C. C., Campbell, J. S., Tonge, R. P., Chen, W., Fausto, N., Nelson, S. D., and Bruschi, S. A. (
Prieto-Alamo, M. J., and Laval, F. (
Saitoh, S., Momoi, M. Y., Yamagata, T., Mori, Y., and Imai, M. (
Sassa, S., and Kappas, A. (
Schierbeek, H., and Berger, R. (
Schulze, A., Frommhold, D., Hoffmann, G. F., and Mayatepek, E. (
Stacpoole, P. W., Henderson, G. N., Yan, Y., Cornett, R., and James, M. O. (
Tao, L., Yang, S., Xie, M., Kramer, P. M., and Pereira, M. A. (
Tong, Z., Board, P. G., and Anders, M. W. (
Tong, Z., Board, P. G., and Anders, M. W. (
Tuchman, M., Whitley, C. B., Ramnaraine, M. L., Bowers, L. D., Fregien, K. D., and Krivit, W. (
Tzeng, H.-F., Blackburn, A. C., Board, P. G., and Anders, M. W. (
Uden, P. C., and Miller, J. W. (
Wyss, P. A., Boynton, S., Chu, J., and Roth, K. S. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments